INTRODUCCIÓN
La creciente presión de la agricultura, así como la búsqueda de una mayor eficiencia en los sistemas ganaderos, ha conducido a un mayor uso de herramientas farmacológicas como estrategias para incrementar la rentabilidad. En ese contexto, el uso de antibióticos ha generado una mejora sustancial de la eficiencia en los sistemas de engorde, hasta alcanzar un uso casi masivo a lo largo de los últimos años 2 . Uno de ellos es la monensina (MN), un antibiótico del tipo poliéter (PE) con actividad como ionóforo, elaborado por el hongo Streptomyces cinnamonensis, utilizado desde hace más de tres décadas como regulador del metabolismo ruminal 1 .
El uso de MN se ha incrementado en los últimos años, al descubrirse sus efectos benéficos sobre la cría vacuna, así como la optimización de la conversión de alimentos y la prevención de patologías como meteorismo, acidosis y coccidiosis bovina. La dosis terapéutica establecida para MN es de 1 a 3 mg/kg de peso vivo (equivalente a 30 mg/kg de alimento). No obstante, con frecuencia se reportan intoxicaciones a causa del uso de formulaciones inadecuadas, incorrecta preparación en la pre-mezcla, fallas en el mezclado y mala distribución en el comedero 7 .
Por estas razones, la búsqueda de un método de cuantificación de MN en piensos, utilizando equipamientos y métodos al alcance de los veterinarios de la especialidad, constituye un objetivo de gran aplicabilidad en producción animal. En el presente trabajo se describe un método analítico concebido para ser desarrollado en laboratorios de baja complejidad y se evalúa su desempeño en diferentes matrices alimentarias.
MATERIAL Y MÉTODOS
Técnica propuesta. Se empleó una modificación del método reportado por el Centro de Inspección de Alimentos y Materiales Agrícolas (FAMIC: Food and Agricultural Materials Inspection Center, Tokyo, Japon) 4 . Tal institución recomienda un proceso que incluye dos etapas, que aquí denominaremos “pre-analítica” y “analítica”. La primera incluye la preparación de la muestra, la obtención de un extracto y la derivatización por copulación con el cromóforo p-dimetil-amino-benzaldehído (PDB). La segunda fase es la determinación espectrofotométrica del producto de la copulación del compuesto, que varía dentro de la escala del color azul, con máxima absorbancia a 578 nm.
Toma de muestras. Se analizaron muestras que contenían diferentes concentraciones de MN conocidas e incógnitas, recolectadas en las provincias de La Pampa y Córdoba, Argentina. Se procesaron diez muestras correspondientes a raciones administradas a bovinos en establecimientos con y sin problemas de intoxicación, así como también muestras de núcleos minerales y vitamínicos con el aditivo de MN. En aquellos casos en los cuales se reportó intoxicación alimentaria, se tomaron cinco alícuotas de distintos lugares del comedero, con posterior homogeneización de las mismas para conformar un volumen final de 500 g. Posteriormente se realizó el muestreo por cuarteo5 y se obtuvo una alícuota final de 100 g.
Procesamiento de la muestra. Las muestras fueron procesadas en un molinillo para reducirlas a un polvo fino. Diez gramos de este material triturado fueron disueltos en 100 ml de etanol anhidro, agitando la mezcla durante 20 minutos para promover una completa homogeneización. Luego, para favorecer la disolución, se sometió la mezcla a agitación/vibración en un baño ultrasónico durante 5 minutos y se filtró para obtener un extracto etanólico. Considerando que el medio acuoso interfiere en la reacción de copulación, todo el proceso se desarrolló evitando la contaminación con agua (reactivos, material de vidrio, operaciones de trasvase). Para establecer la curva de calibración se dispuso de un pienso como matriz de referencia. A partir de ella se preparó una muestra de referencia por incorporación de cantidades apropiadas de MN de calidad analítica. Se prepararon dos extractos etanólicos, uno desde la matriz de referencia (extracto A) y otro desde la muestra de referencia (extracto B). A partir de estos dos extractos se preparó una serie de diluciones en el rango de 0 a 110 mg MN/kg de alimento con cinco niveles, por triplicado.
Reacción de derivatización.La reacción se efectuó en medio ácido, garantizado por la adición de H2 SO4 . El reactivo derivatizante fue el PDB, que generó un producto de color azul, con un máximo de absorción a 578 nm. Se preparó PDB en etanol anhidro (1,2 mg/ ml), con un contenido final de 1% de H2 SO4 98%. Esta solución fue preparada el mismo día de trabajo (no puede almacenarse). Se dispuso de una batería de tubos de ensayo por triplicado, en cada uno de los cuales se colocaron 3 ml de la matriz de referencia (extracto A) con la concentración de MN correspondiente a cada punto de la curva, más 1 ml de la solución de PDB. Todos los tubos fueron tapados e incubados en baño termostático a 70ºC durante 20 min para el desarrollo de la reacción.
Espectrofotometría.Una vez frías, las soluciones se analizaron en un espectrofotómetro a 578 nm, utilizando como blanco de reactivo una solución de etanol: H2 SO4 98%. En las muestras que registraron concentraciones superiores a las del rango establecido por la curva de calibración, las lecturas fueron repetidas previa dilución al 1/2 con el extracto A.
Patrón secundario. El patrón de referencia para la puesta a punto del método fue una sal sódica de MN (Sigma-Aldrich). Para establecer la curva de calibración se utilizó un extracto obtenido por cristalización proveniente de concentrados minerales comerciales de MN de uso veterinario. Esta MN fue comparada mediante espectroscopía infrarroja con la sustancia de referencia, y debió demostrar características analíticas equivalentes a las obtenidas con el patrón comercial.
RESULTADOS
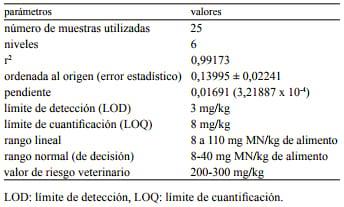
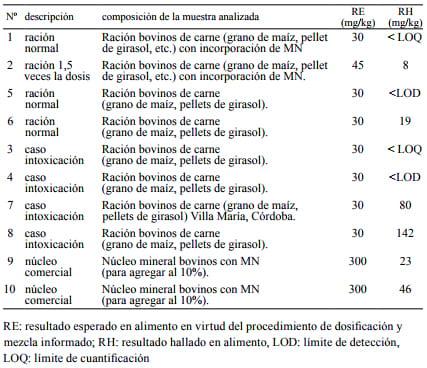
En la Tabla 1 se presentan los datos correspondientes a la curva de calibración de MN ajustada por regresión lineal. En la Tabla 2 se detallan las muestras analizadas y la composición de cada una de ellas, así como los resultados esperados y obtenidos.
En piensos formulados bajo diferentes condiciones (no asociados a casos de intoxicación) se registraron valores en un rango que iba desde el límite de detección de la técnica hasta 19 ppm. En muestras obtenidas en casos de intoxicación se hallaron valores que se ubicaron por debajo del límite de detección hasta niveles de 142 ppm. En muestras provenientes de núcleos comer ciales, donde se esperaban resultados 10 veces mayores a los del pienso (300 ppm) se hallaron niveles máximos de 46 ppm.
Tabla 1. Evaluación estadística del método analítico.
Tabla 2. Concentración de monensina esperada versus hallada en distintas muestras de alimentos.
DISCUSIÓN
En la región denominada “pampa húmeda” de Argentina, se han reportado varios incidentes aislados, relacionados con la intoxicación del ganado por sobredosificación con MN. En ninguno de dichos casos se realizó la determinación de la concentración de MN en los piensos involucrados. Ello fue así fundamentalmente porque no existen protocolos de preparación de piensos adicionados con premezclas conteniendo MN, destinados al control de las concentraciones finales del antibiótico. Tal afirmación se basa en nuestras experiencias adquiridas en el campo y coinciden con afirmaciones publicadas por otros colegas 10 .
La MN posee una gran afinidad por cationes como el sodio, lo cual altera in vivo la polaridad de la membrana celular, afectando las fibras de los músculos esquelético y cardíaco, generando una miocardiopatía que cursa con estasis sanguínea y consecuente edema intersticial pulmonar 8 . La toxicidad puede presentarse tanto en forma aguda como crónica, dependiendo de la dosis ingerida. La DL50 en vacunos se ha establecido entre 50 y 80 mg/kg de peso vivo, en tanto que las primeras manifestaciones clí- nicas de intoxicación aparecen frecuentemente con dosis de 10 mg/kg de peso vivo 9 .
Ello implicaría un estrecho margen para las sobredosis, con elevado riesgo de muerte del animal intoxicado. Sin embargo, las tasas de mortalidad reportadas han sido muy variables con valores que oscilan entre 1% y 26% 7, 10, 11 . La ausencia de acuerdo respecto a los márgenes de seguridad de MN puede ser una de las causas que está generando un creciente aumento de casos de intoxicación en bovinos bajo alimentación controlada.
A comienzos de la década de 1970 se establecieron varios métodos analíticos de detección y confirmación de uno o más antibióticos del tipo PE en alimentos, tal como la MN. La mayoría de tales métodos se basan en cromatografía líquida de alta presión (HPLC), lo cual requiere un equipamiento de laboratorio de alta complejidad y -consecuentemente- elevado costo. Dado que varios de estos PE no responden al ultravioleta o a la fluorescencia, los procedimientos analíticos deben recurrir a pasos previos denominados “derivatizaciones” o al uso de detectores de masa, lo cual torna más complejas las mediciones 3, 4, 6 .
En cuanto al método aquí descrito, aunque parcialmente laborioso, permite obtener resultados certeros dentro de los rangos de interés esperados. La variabilidad en cuanto a los resultados obtenidos, no solo para aquellos casos en los cuales la concentración de MN se encontraba dentro de los límites terapéuticos, sino también para aquellas muestras correspondientes a casos de intoxicación, pudo ser ocasionada por fallas del mezclado o una muestra de pienso que no correspondió al momento en que se presentó el caso.
Este punto parece ser crítico a la hora de evaluar la concentración de MN, dado que el proceso analítico mostró muy buena respuesta en la prueba de calibración. En establecimientos de gran envergadura, las dietas son preparadas diariamente en el mixer (carro de mezclado del alimento), por lo cual la muestra tomada para el análisis, puede tener una composición tal vez diferente a la que generó el caso de intoxicación, arrojando un resultado falso negativo. Asimismo, debe tenerse en cuenta que cuando aparecen los signos de intoxicación y/o muertes, muchas veces son causados por alimentos administrados horas o días previos al momento del muestreo.
Los ensayos preliminares indican que esta técnica es útil para la detección de MN en piensos, aunque no para otras formulaciones como pre-mezclas y alimento balanceado, considerando que ciertos componentes del alimento podrían generar un efecto matriz que dificulte la interpretación de los resultados. Del presente trabajo surge la necesidad de evaluar un mayor número de muestras, así como la necesidad de diseñar y aplicar muestreos representativos del comedero que uniformicen tanto la cantidad como la calidad de las muestras, para obtener alícuotas representativas de la partida de alimento problema.
A manera de conclusión es dable resaltar que la técnica ensayada en piensos se reveló útil para evaluar MN tanto en dosis terapéuticas como en sobredosificaciones. Aunque requiere cierta laboriosidad, es un método de bajo costo en cuanto a insumos y equipamientos requeridos, comparado con otros procedimientos. Su aplicación permitirá optimizar la formulación de las dietas y la concentración de sus componentes, evitando trastornos clínicos y subclínicos capaces de afectar la rentabilidad del sistema ganadero. Asímismo, tal práctica contribuiría a regular el uso racional de los productos farmacológicos de uso veterinario.
REFERENCIAS
- Bogaert C, Jouany JP, Jeminet G. 1990. Effects of the ionophore antibiotics monensin, monensin-propionate, abierixin and calcimycin on ruminal fermentations in vitro. Anim Feed Sci Technol 28: 183-197.
- Bretschneider G. 2009. Beneficios del uso de monensina en la alimentación del ganado para carne, leche y cría. Rev Electr Vet ISSN 1695-7504, vol. 10, Nº 10.
- Dusi G, Gamba V. 1999. Liquid chromatography with ultraviolet detection of lasalocid, monensin, salinomycin and narasin in poultry feeds using pre-column derivatization. J Chromat A, 835: 243-246.
- Food and Agricultural Materials Inspection Center (FAMIC). 2013. Manual sobre métodos de determinación de monensina sódica. Disponible on line: www.famic. go.jp/ffis/oie/obj/a28_mn.pdf , acceso 20-04-2013.
- Greenfield H, Southgate DA. 2006. Datos de composición de alimentos: obtención, gestión y utilización, Public. FAO, Roma, 2da ed., p. 238.
- Nebot C, Iglesias A, Regal P, Miranda JM, Fente C, Cepeda A. 2012. A sensitive and validated HPLC–MS/ MS method for simultaneous determination of seven coccidiostats in bovine whole milk. Food Control 27: 29-36.
- Odriozola N. 2004. Intoxicación por monensina. Diagnóstico Veterinario Especializado. INTA EEA Balcarce, Argentina. Disponible on line: http://www. produccionanimal.com.ar/sanidad_intoxicaciones_metabolicos/ intoxicaciones/41-i ntoxicacion_por_monensina.pdf]. Acceso: 24-09-15.
- Pascuet ML, Moore DP, Iraguen I, Cosentino IA, Odriozola E. 2005. Cambios enzimáticos, lesiones macroscópicas y microscópicas producidas por la intoxicación con monensina en bovinos. Rev Med Vet 86: 47-51.
- Redostits OM, Gay CC, Blood DC, Hinchcliff KW. 2002. Medicina Veterinaria (Capítulo: Intoxicación por ionóforos), 9º ed., McGraw-Hill, Madrid, p.1931.
- Rodríguez Armesto R, Peralta C, Ochoteco M, Picco E, Litterio N, Boggio JC. 2004. Posible intoxicación accidental con monensina en terneros destetados. Vet Arg 201: 13-20.
- Vickovic I, Sostaric B, Toncic J. 2010. Monensin toxicity and acute lethal rhabdomyolysis in accidental exposure in cattle (Abstracts of the XII International Congress of Toxicology), Toxicol Lett 196: 17, pag. S233.







.jpg&w=3840&q=75)