Las enfermedades Prion o Encefalopatías Espongiformes Transmisibles
Publicado: 1 de enero de 2002
Por: Carlos Sánchez Huertas
Introducción
Prion: naturaleza del agente infeccioso.
Prion: naturaleza del agente infeccioso.
La función del prion.
La bivalencia del prion.
La infección.
Nociones sobre estructura del prion.
La barrera de las especies.
La diversidad de los priones.
Nomenclatura.
Estructura y expresión del gen PrP.
Prevención y tratamiento.
La herencia de la levadura.
El doble juego de una proteína
Bibliografía
La bivalencia del prion.
La infección.
Nociones sobre estructura del prion.
La barrera de las especies.
La diversidad de los priones.
Nomenclatura.
Estructura y expresión del gen PrP.
Prevención y tratamiento.
La herencia de la levadura.
El doble juego de una proteína
Bibliografía
Introducción.
El término "prion" es usado para describir el agente infeccioso responsable de varias enfermedades neurodegenerativas encontradas en los mamíferos. La palabra en sí deriva de "proteinaceous infectious particle", definición propuesta porStanley B. Prusiner.
Esta definición vino dada por la hipótesis inicial, que más tarde se confirmaría, de que este agente infeccioso consistía únicamente en una proteína, carente de genoma y ácidos nucleicos.
Se ha observado esta proteína en las membranas neuronales de los mamíferos sin causar enfermedad alguna, pero se sabe que un cambio conformacional de su estructura terciaria puede provocar la aparición de la enfermedad. Estas proteínas en su forma patógena se multiplican exponencialmente al ponerse en contacto con las proteínas normales, ya que les inducen el cambio conformacional que las vuelve infecciosas.
La aparición de estos desordenes estructurales en las proteínas, pueden ser transmisibles, heredados, o incluso esporádicos, es decir, sin evidencias de transmisión ni herencia.
Las enfermedades prion (colectivamente llamadas "encefalopatías espongiformes transmisibles") conocidas hasta ahora son fatales, afectan al sistema nervioso y se cree que también a los músculos. Estas enfermedades pueden incubarse durante años o incluso décadas en humanos, de ahí que inicialmente se las conociera como "virus lentos".
Arriba
Prion: naturaleza del agente infeccioso.
Se han realizado multitud de experimentos para demostrar la naturaleza de los priones:
| - Filtrable con poros 25 nm o 100 nm. - Es invisibles al microscopio óptico y electrónico. - Resistente a: EDTA Proteasas ( Tripsina, pepsina ), aunque reducen la infectividad Nucleasas ( ribonucleasas A y III, desoxiribonucleasa I ) Calor ( 80ºC ) Radiación ultravioleta ( 2540 Å ) Radiación ionizante Psoralenos e iones Zn | - Largo periodo de incubación ( meses, años, décadas). - No producen respuesta inflamatoria - No antigénicos. - Patología crónica progresiva. - Fatal en todos los casos. - Carecen de cuerpos de inclusión. - Presencia de ácido nucleico no demostrada. - El único componente conocido es la proteína - PrP. - Pueden existir en múltiples formas moleculares - Periodo de adaptación a nuevos hospedadores. - Control genético de la susceptibilidad de algunas especies. - Existencia de distintas cepas. |
Métodos de Inactivación : |
- Autoclave >134 °C
- Hipoclorito sódico (20ºC, 1hora) - Hidroxido sódico 2N - Fenol 90 - Eter - Acetona - Permanganato potásico 0.002 M - Urea 6 M - 2-Cloroetanol - Cloroformo |
A pesar de los grandes esfuerzos realizados para encontrar el material genético de este agente infeccioso, solo pequeños fragmentos de menos de 100 nucleótidos han permanecido después de los procesos de purificación (Meyer 1991), sospechándose que pueda ser únicamente material contaminante.
Tras descartar la presencia de ácidos nucleicos en los priones, también se desestimaron los carbohidratos y lípidos como elementos infecciosos. Prusiner resume que solo los procedimientos que hidrolizan o afectan a proteínas logran modificar la intensidad de la infección.
Se ha asumido la presencia de un pequeño ligando unido al prion, como componente esencial de la partícula infecciosa, ya que no se ha podido eliminar.
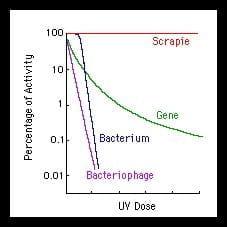
Hipótesis Virales
Hipótesis Virales
A pesar de que no se han encontrado pruebas concluyentes de la existencia de ácidos nucleicos asociados con el prion, algunos investigadores creen que los priones se forman cuando la proteína prion se asocia con un ácido nucleico patógeno extraño.
Gajduseky cols. apuestan por la teoría vírica, ya que los viroides han demostrado resistencia a la radiación ultravioleta de 254 nm, al igual que la partícula PrP, que pese a mantener su carácter infeccioso a temperaturas superiores que lo agentes infecciosos convencionales, observó que eran rápidamente inactivados al sobrepasar los 85ºC.
C. Weissmannpor su parte, propone que el agente está formado por dos componentes:
- El PrPSc (apoprion), que puede causar enfermedades transmisibles incluso libre de ácidos nucleicos.
- Un ácido nucleico (coprion), del cual pueden existir muchas variantes. Es el que determina las propiedades fenotípicas que definen el linaje del agente infecciosoAmbas partes constituyen lo que él denominó holoprion.
- El PrPSc (apoprion), que puede causar enfermedades transmisibles incluso libre de ácidos nucleicos.
- Un ácido nucleico (coprion), del cual pueden existir muchas variantes. Es el que determina las propiedades fenotípicas que definen el linaje del agente infecciosoAmbas partes constituyen lo que él denominó holoprion.
Laura Manuelidis,de la Universidad de Yale (EEUU) afirma que existen virus que disponen de un sistema de reparación del material genético que les permite resistir radiaciones similares a las que resiste el PrPSc; así como virus lentos convencionales, que escapan al sistema inmunitario instalándose en el interior de las células.
L. Manuelidis hace especial hincapie en varias características de los agentes infecciosos, a saber:
- La existencia de distintas cepas o razas de priones que causan diferentes patrones de enfermedad. Solo pueden aparecer representados en diferentes linajes o razas, aquellos patógenos que poseen ácidos nucleicos.
- Su multiplicación exponencial.
- Su tiempo de latencia prolongado.
- Infección del sistema retículo-endotelial ( bazo, glóbulos blancos ).
La evidencia de que la enfermedad nvCJD procedía de la epidemia de las vacas locas (BSE) constituyó otro argumento a favor, ya que la transmisión por vía oral pone muy difícil la resistencia de un agente puramente proteico a la acidez y las enzimas del aparato digestivo. En cambio, muchos virus sí son capaces de sobrevivir a estas condiciones.
- La existencia de distintas cepas o razas de priones que causan diferentes patrones de enfermedad. Solo pueden aparecer representados en diferentes linajes o razas, aquellos patógenos que poseen ácidos nucleicos.
- Su multiplicación exponencial.
- Su tiempo de latencia prolongado.
- Infección del sistema retículo-endotelial ( bazo, glóbulos blancos ).
La evidencia de que la enfermedad nvCJD procedía de la epidemia de las vacas locas (BSE) constituyó otro argumento a favor, ya que la transmisión por vía oral pone muy difícil la resistencia de un agente puramente proteico a la acidez y las enzimas del aparato digestivo. En cambio, muchos virus sí son capaces de sobrevivir a estas condiciones.
Mutaciones y polimorfismos del gen PRNP podrían inducir mayor susceptibilidad del huésped frente a ciertas cepas del agente infeccioso.
Pruebas
H. Diringerhalló en 1994 lo que parecía ser la primera evidencia física de la existencia de los virus. Su tamaño era inesperadamente pequeño : 10 a 20 nm. Un año después, el equipo de Manuelidis obtuvo un gel de electroforesis con bandas que revelaban la existencia de RNA en extractos cerebrales de hamsters con CJD. Las bandas no pudieron localizarse en cerebros no infectados ni en las librerias genómicas.
El tamaño de la partícula infecciosa fue evaluado en unos 27 nm, tamaño similar al de varios virus conocidos. El genoma se estimó en 1.000-3.000 pares de bases.
El tamaño de la partícula infecciosa fue evaluado en unos 27 nm, tamaño similar al de varios virus conocidos. El genoma se estimó en 1.000-3.000 pares de bases.
Los investigadores concluyen que un ácido nucleico y una o varias proteínas constituyen los componentes intrínsecos del virus CJD (1). Los dos componentes no son infecciosos, a menos que se asocien en partículas nucleasa-resistentes.
La función del prion.
Los Los priones son proteínas observadas comúnmente en la superficie de las neuronas de todos los mamíferos estudiados. La función que cumple esta proteína ha sido estudiada por un grupo de investigadores japoneses liderados por Suchiro Sakaguchi, de la Universidad de Nagasaki. Se crearon ratones homozigotos respecto a la falta del gen PRNP, que codifica para la isoforma normal del prion (PrPc).(2)
Estos ratones son normales hasta las 70 semanas de vida, a partir de aquí comenzaron a manifestar importante pérdida de coordinación, temblores al andar, incapacidad de mantener una trayectoria; a las 90 semanas, incapacidad de mantenerse y movimientos espasmódicos en sus patas traseras, muchos de ellos tenían la columna vertebral arqueada con una convexidad hacia atrás.
Fisiológicamente, el cerebelo (encargado de la coordinación) se había encogido hasta un tercio respecto al de los ratones normales, y había una grave deficiencia de las neuronas de Purkinje. Estas células son una variedad de neurona que constituye el elemento fundamental del córtex cerebeloso. El equipo investigador observó que los niveles de las células de Purkinje eran normales en los ratones jóvenes, así que afirmó que estas células podían sobrevivir cierto periodo de tiempo sin ayuda, pero morían al poco tiempo sin la presencia del producto del gen PrP.
Al inocular los ratones nulos para el gen PRNP (por tanto sin la forma natural PrPc) con una partícula prion infecciosa (PrPSc), no desarrollaban la enfermedad (Beler 1992) y no se detectaron evidencias de replicación del PrPSc. De esto se dedujo que, para causar la enfermedad era necesaria la presencia del tándem PrPc-PrPSc.(3)
Este experimento parecía esclarecer un poco la función de la proteína prion, ya que la ausencia (sea por ausencia del gen o transformación de la forma natural a la infecciosa) tiene ciertas consecuencias concretas en el individuo. Se le podría atribuir un papel en la supervivencia de las neuronas de Purkinje en el cerebelo.
Sin embargo posteriormente, se han creado nuevamente modelos de ratón carente del gen PRNP que han crecido y se han desarrollado de una manera normal, con algunas excepciones de ataxia y alteración del ritmo circadiano más allá de los dos años de edad (Beler 1992, Tobler 1996, Prusiner 1998).
La función de la forma celular del prion es hoy aun desconocida, aunque se barajan varias:
- Proteína de transducción de señal (4)
- Adhesión celular
- Superóxido dismutasa (11)
- Regulación y distribución de los receptores de acetilicolina
- Proteína de transducción de señal (4)
- Adhesión celular
- Superóxido dismutasa (11)
- Regulación y distribución de los receptores de acetilicolina
Arriba
4
La bivalencia del prion.
La naturaleza del prion le permite manifestarse de 2 formas: una forma celular o PrPc de 33-35 KDa y otra patógena PrPSc o scrapie PrP de 33-35 Kda. El tratamiento de esta última con proteinasa K nos da una tercera forma de 27-30 Kda que mantiene intacta su capacidad infecciosa y es el núcleo proteasa resistente.
La PrPc es una proteína de membrana, la PrPSc 33-35 se acumula dentro de la célula con posibilidades de agregación, la PrPSc 27-30 se acumula extracelularmente y puede formar placas. El test de detección "Prionics" se basa sencillamente en detectar mediante electroforesis la presencia de PrPSc 27-30 en preparados cerebrales.
Las formas PrPc y PrPSc lo son de la misma proteína, es decir, comparten la misma secuencia de aminoácidos (estructura primaria); se comprobó la imposibilidad de que las dos isoformas PrP fueran productos de dos tipos de ARNm distintos. La diferencia entre ambos reside en su estructura secundaria y terciaria, es decir, en su conformación espacial. La forma PrPSc ha sufrido un plegamiento respecto la forma celular PrPc.
El plegamiento de las formas nativas está inducido por partículas PrPSc infecciosas que ponen en marcha una reacción en cadena, de forma que la infección se desarrolla exponencialmente.
Las variantes familiares de estas enfermedades (que se transmiten por herencia familiar) demuestran la existencia de un factor genético que contradice en cierto modo la teoría de secuencia idéntica de las 2 formas. Se compararon clones del gen PrP procedentes de un paciente con la enfermedad de Gerstmann-Sträussler-Scheinker (GSS) con genes PrP obtenidos de la población sana, y se encontró que un par de bases (de un total de 750) era diferente. Este cambio en el codón 102 (P102L), era responsable de la sustitución del aminoácido nativo prolina (Pro) por leucina (Leu) en la proteína PrPc del paciente. Esta sustitución sería la responsable de la desestabilización de la estructura terciaria de la proteína y su transformación en agente patógeno.
Así pues, determinadas mutaciones en el gen causan susceptibilidad a la enfermedad.
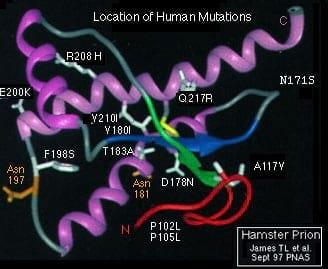
Las conexiones genéticas introducen la posibilidad de que estas enfermedades puedan aparecer sin infección, incluso también sin que exista una relación hereditaria obligada, ya que estas mutaciones pueden aparecer espontáneamente, producir el consiguiente cambio conformacional y causar la enfermedad. Las demencias por priones esporádicas son las más comunes (aproximadamente el 85% de los casos), la CJD esporádica (129 M/M) es la más común de todas ellas.
La Enfermedad Heredada
La Enfermedad Heredada
Alrededor del 15% de las demencias por prion son heredadas o tiene agregación familiar. El patrón de transmisión de las enfermedades prion familiares (o heredadas), se reconoce como autosómico y dominante.
Las proteínas procedentes de genes PRNP mutados no adoptan su forma patógena nada más sintetizarse, sino más tarde. Esto es porque las mutaciones del gen PRNP hacen las proteínas PrPc más susceptibles a plegarse, pero llevaría cierto tiempo hasta que estas moléculas lo hicieran espontáneamente y se iniciara la enfermedad.
Inicialmente se asociaron tres enfermedades con mutaciones en el gen PRNP:
- La enfermedad de Creutzfeldt-Jakob familiar (fCJD).
- El síndrome de Gerstmann-Stréessler-Scheinker (GSS).
- El insomnio fatal familiar (IFF).
Posteriormente se han asociado significativamente hasta 13 mutaciones del PRNP con el desarrollo de la enfermedad (Prusiner 1994).
- La enfermedad de Creutzfeldt-Jakob familiar (fCJD).
- El síndrome de Gerstmann-Stréessler-Scheinker (GSS).
- El insomnio fatal familiar (IFF).
Posteriormente se han asociado significativamente hasta 13 mutaciones del PRNP con el desarrollo de la enfermedad (Prusiner 1994).
Al fCJD se ha asociado una mutación en el codon 129 del gen PRNP que cambia meteonina por valina y, según los polimorfismos del gen, predispone para unos síntomas u otros. El GSS está asociado a una mutación en el codon 102 que cambia leucina por prolina, aunque se han asociado al menos tres mutaciones más. El IFF se asocia a la mutación puntual en el codon 178 que cambia el ácido aspártico por la asparragina. Esta información está muy resumida.
Igualmente se ha demostrado la conexión genética con otras 4 mutaciones p.ej: fCJC (E200K); las mutaciones artificiales A113V, A115V y A118V inducen al plegamiento de la proteína PrPc y a la producción de la enfermedad. Lo que demuestra la relevancia del aminoácido Val en el mantenimiento de la estructura de la PrPc.
Arriba
La infección.
Para comprender mejor este apartado debería tratarse en dos puntos:
- La aparición de la partícula infecciosa en el huésped.
- El desarrollo de la enfermedad o proliferación del patógeno.
En el primer apartado deben distinguirse tres orígenes de infección: iatrogénico (transmitido), heredado y la mutación espontánea. Son las tres formas de aparición de la primera partícula infecciosa: procedencia externa, mutación heredada del gen PrP y mutación esporádica de dicho gen.
- La aparición de la partícula infecciosa en el huésped.
- El desarrollo de la enfermedad o proliferación del patógeno.
En el primer apartado deben distinguirse tres orígenes de infección: iatrogénico (transmitido), heredado y la mutación espontánea. Son las tres formas de aparición de la primera partícula infecciosa: procedencia externa, mutación heredada del gen PrP y mutación esporádica de dicho gen.
La multiplicación del agente infeccioso es exponencial. La isoforma patógena es captada por fagocitosis en neuronas o glia, y transportada al lisosoma para su degradación, en el lisosoma se produce contacto entre PrPSc y PrPc y la primera induce el plegamiento de la segunda. Las PrPSc (el núcleo 27-30 KDa) son relativamente resistentes a las proteasas y se acumulan, los lisosomas revientan cuando se supera un determinado volumen, liberando al citosol las PrPSc y proteínas hidrolíticas que contenían. Las proteínas hidrolíticas degradan la célula, las PrPSc (en su forma 27-30) quedan libres en el espacio extracelular, se agregan y forman placas amiloides. El proceso se repite en las células adyacentes, creando agujeros en el tejido cerebral.
Otra teoría trata de explicar el desarrollo de la patología, es la Teoría Cristalina, basada en que la PrPSc forma estructuras cristalinas muy insolubles, que pueden ser inducidas en cualquier momento del ciclo de la PrPc.
Estas enfermedades neurodegenerativas se caracterizan por una tríada patológica:
- Vacuolización o espongiosis, en el citoplasma de neuronas y células gliales. En las enfermedades de Alzheimer o Huntington, la vacuolización es en el espacio extracelular como resultado de la pérdida de neuronas.
- Reacción glial o gliosis (crecimiento exagerado de las células gliales) en ausencia de cambios infamatorios.
- Pérdida de neuronas.
Estos síntomas pueden observarse en cualquier área y lámina del neocórtex, en el putamen, núcleo caudado, tálamo, capa molecular de la corteza cerebelosa; y son mínimos o ausentes en el hipotálamo, globus pallidus, talo cerebral y espina dorsal. La sustancia blanca no presenta cambios patológicos (daSilva 1994).
- Vacuolización o espongiosis, en el citoplasma de neuronas y células gliales. En las enfermedades de Alzheimer o Huntington, la vacuolización es en el espacio extracelular como resultado de la pérdida de neuronas.
- Reacción glial o gliosis (crecimiento exagerado de las células gliales) en ausencia de cambios infamatorios.
- Pérdida de neuronas.
Estos síntomas pueden observarse en cualquier área y lámina del neocórtex, en el putamen, núcleo caudado, tálamo, capa molecular de la corteza cerebelosa; y son mínimos o ausentes en el hipotálamo, globus pallidus, talo cerebral y espina dorsal. La sustancia blanca no presenta cambios patológicos (daSilva 1994).
Las vacuolas tienen un tamaño entre 20 y 200 micras, pueden agregarse formando grandes vacuolas que distorsionan la citoarquitectonia. Aparecen en el citoplasma de las neuronas de cada capa de la corteza cerebral, también en neuronas de la corteza.
La espongiosis cortical puede ir acompañada de procesos análogos en ganglio basal, tálamo y corteza cerebelosa.
Se observan depósitos amiloides de PrPSc 27-30 (insolubles) en forma de placas, llamados placas amiloides, que se tiñen positivas para la PrP (al contrario de las placas presentes en el Alzheimer). Tienen afinidad por la eosina, rojo congo. Presentes sobretodo a nivel del cerebelo y, minoritariamente, a nivel del tálamo, ganglios basales y corteza cerebral. Son un buen indicador de la infección, pero aparentemente no son causa importante de la enfermedad.
Estudios ultraestructurales han demostrado una relación de la microglía con la formación de estas placas.
Se ha comprobado que las moléculas de PrPc se localizan en lugares distintos a los que se encuentran las partículas PrPSc. La materia blanca, ovillos de axones mielinizados, contienen PrPSc pero están libres de PrPc. Estas y otras investigaciones demuestran que los priones se mueven a través de los axones de las neuronas, y la infectividad de PrPSc se basa en un patrón de transporte retrógradoa lo largo del axón.
Todos estos síntomas aparecen sin que se produzca respuesta inmune, a excepción de un aumento en los niveles de citokinas en los estados más tardíos de la enfermedad.
Los priones pueden ser transmitidos de un ser vivo a otro, enfermedades de origen iatrogénico (enfermedad del Kuru, CJD iatrogénico) de forma comprobada inoculándolos directamente al cerebro, la piel o el tejido muscular; y también a través de la alimentación (nvCJD, que es un tipo de iatrogénica).
En experimentos se ha detectado la presencia del agente prionico en la sangre, pero no se han dado casos reales de transmisión de la enfermedad por transfusiones en humanos. Aunque sí se ha descubierto que el BSE puede ser transmitido en ovejas por transfusiones de sangre de donantes que aun no hayan manifestado la enfermedad (12).
Por todo esto, ya no se usa sangre completa para transfusiones en el Reino Unido, sino que se le extraen todos los glóbulos blancos en un proceso llamado leucodepletion, además, ha cesado la producción de productos derivados de la sangre donada por británicos, ahora se importa plasma de otros países con baja incidencia de BSE y ausencia de nvCJD
La Infección por BSE
La Infección por BSE
Los priones ingeridos pueden ser absorbidos por los parches de Peyers del intestino; éstos son parte del MALT o tejido linfoide asociado a mucosa. Se cree que MALT presenta microorganismos al sistema inmune, facilitando la respuesta inmunitaria frente a éstos. Los priones serían captados del mismo modo.
Depués de fagocitar la partícula, las células linfoides viajan a otros tejidos linfáticos, como nódulos linfáticos, bazo y las amígdalas; el prion podría replicarse en estos lugares. Muchos de estos lugares están inervados y el prion puede acceder a un nervio, ascender retrógradamente por el axon a la médula espinal y finalmente al cerebro.
Para que una infección desde tejidos periféricos tenga éxito, deben expresar el gen PRNP, además de neuronas del Sistema Nervioso Central, células del Sistema Linfoide y neuronas periféricas. Los linfocitos B maduros también son necesarios para el desarrollo de la enfermedad partiendo de una ruta periférica.
Tanto neuronas como células gliales pueden propagar la enfermedad independientemente.
Astrocitos y otras células gliales presumiblemente, tienen un papel en la patogénesis, dado que retrovirus que infectan la glia, producen degeneración espongiforme del cerebro.
En el caso particular de la enfermedad iatrogénica por ingestión de material de riesgo infectado con BSE (nvCJD) plantea numerosas dudas:
¿Cómo una proteína sobrevive a los ácidos digestivos?
¿Cuánta cantidad de prion patógeno hay que ingerir para que aparezca la enfermedad? ¿Cuánto tarda en aparecer?
¿Cuánta importancia tiene la predisposición genética?
¿Cómo llega el prion patógeno al tejido nervioso?
Dado que el músculo recibe inervación ¿es peligroso comer tejido muscular?
Está demostrado que la nueva variante de la enfermedad Creutzfeldt-Jakob (nvCJD) está causada por el agente responsable del BSE en bovinos.(5)
Si comemos material de riesgo infectado por BSE existe probabilidad de que nos contagiemos de la enfermedad, esa probabilidad viene determinada por varios factores: la predisposición genética y la cantidad de material de riesgo que comamos.
Todas las proteínas de nuestra dieta, son despiezadas a aminoácidos en la digestión para incorporarse a la síntesis proteica u otras rutas metabólicas. Como en todo, existe una mínima probabilidad de que esto no ocurra, y una cantidad de proteínas sobreviva a los ácidos y sea absorbida en el intestino. La probabilidad de que esto ocurra es mínima, pero será mayor cuanta más cantidad de proteína hayamos comido.
La predisposición genética de un ser humano a padecer nvCJD radica en el codon 129 del gen PRNP. Todos los casos de nvCJD estudiados eran homocigotos para Meteonina en este codon (M/M 129), por lo tanto, personas homocigotas para Valina en este codon (V/V 129) o heterocigotas (M/V 129) pueden ser relativamente resistentes a la infección, tener tiempos de incubación más largos o presentar diferentes síntomas.
El tiempo de incubación hasta que se manifiestan los síntomas es variable y depende de los factores anteriormente nombrados. El máximo tiempo registrado está entre los 2-2,5 años.
En cuanto a la ingestión de tejido muscular, el riesgo que representa es ínfimo, ya que es muy poco el tejido nervioso que pueda contener.
Si encontrais algún error o no estais de acuerdo con lo que digo, por favor corregidme mandándome un correo a mi dirección (al final).
Arriba
La estructura del prion.
Estudios mediante los métodos FTIR y CD han mostrado que la proteína PrPc contiene un 40% de hélice-a y muy poca proporción de hoja-b, mientras que la PrPSc se compone de un 30% de hélice-a y un 45% de hoja-b. Pese a esto, ambas proteínas tienen la misma secuencia de aminoácidos.
La proteína PrP está constituida por cuatro regiones de estructura secundaria llamadas H1, H2, H3 y H4, en estas regiones se identifican tres zonas de hélice-a llamadas A, B y C, y dos de hoja-b, llamadas S1 y S2.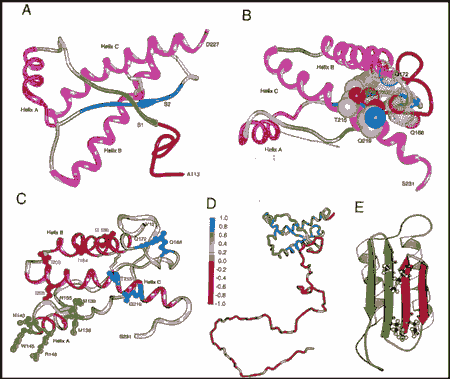
Estructura de la PrPc. B) Lugar donde se cree que la proteína X se liga a la PrPc. C) Lugares que se cree relacionados con la transmisión de priones entre las especies y control de unión de la proteína X D) Flexibilidad de la cadena del polipéptido Prp. E) Modelo para la estructura terciaria de la HuPrPSc.
Se sabe que ambas isoformas PrP poseen "anclas GPI" (glycoinositol phospolipid), gracias a la cual, la PrPc se encuentra "anclada" en la superficie de la célula. Su transformación a PrPSc solo tiene lugar cuando la PrPc alcanza dicho lugar. La PrPSc se encuentra dentro de la célula en estructuras del sistema endocítico y fuera de la célula en placas amiloides como PrPSc 27-30 KDa (Pan 1993).
Los residuos 113-128, dentro de la región H1, son los más conservados en todas las especies estudiadas, y corresponden a una zona transmembranal de la PrPc que se organiza en hélice- a .
El cobre (Cu2+) parece tener un importante papel en la estabilidad de las formaciones en hélice-a. En todas las moléculas de ShaPrP observadas, se han encontrado dos átomos de Cu2+ unidos a ella. Desequilibrios en la homeostasis del cobre conducen a disfunciones del sistema nervioso central (SNC), aunque no está demostrado que este mal funcionamiento sea debido a anormalidades en el metabolismo del PrP.
La adquisición de la resistencia a las proteasas de la PrPSc, es un proceso post-translacional, es decir, se adquiere tras el plegamiento. En el estudio de los modelos de PrPSc se halló un péptido menor derivado de este último, que constituía el núcleo proteasa-resistente de la PrPSc. Este péptido tenía una masa molecular de 27-30 kDa y se le llamo PrP27-30. Se ha encontrado este péptido en todos los casos de enfermedad prion, salvo en las enfermedades del Alzheimer, Parkinson y esclerosis miotrópica lateral.
Estudios sobre el PrPSc proponen que el plegamiento de la PrPc se refiere únicamente al cambio de conformación a hoja-b de la región situada entre los residuos 90 y 140. También el lazo disulfato unido a los extremos COOH de las hélices es necesario para la formación del PrPSc.
La región implicada en la transformación del PrPc en PrPSc se ha situado, aunque no completamente, en el dominio compuesto por los residuos 90-112. Pese a estos resultados, las mutaciones causantes de las enfermedades prion heredadas se localizan a lo largo de toda la proteína.
Estudios con transgenes quiméricos de Sha/Mo y Hu/Mo, identificaron un dominio entre los residuos 95-170 donde el PrPc se une al PrPSc. Posteriormente, se identificó un segundo dominio entre los residuos 180-205 que parecen modular la interacción entre PrPc y PrPSc.
Básicamente puede decirse que la isoforma mutante difiere de la silvestre en:
- Alto contenido en hélices-b .
- Es insoluble en detergentes.
- Es resistente a la proteolisis.
- Localización extracelular.
- Propensión a agregarse
- Alto contenido en hélices-b .
- Es insoluble en detergentes.
- Es resistente a la proteolisis.
- Localización extracelular.
- Propensión a agregarse
Minipriones
Se realizaron diversas pruebas para investigar la naturaleza patógena del prion, y se crearon priones mutantes para facilitar esto. Así, la destrucción de cualquiera de las cuatro regiones en hélice-a de la proteína PrPc, previenen la formación del PrPSc. Mientras que suprimir la región del extremo NH2 con los residuos 23-89, así como otros 36 residuos entre las posiciones 141-176 (ambas regiones en verde: hélice A y hoja S2) no afectan a la aparición de PrPSc.
La molécula PrP resultante con 106 aminoácidos, fue denominada PrP106 (o miniprion). La cual tiene idénticas propiedades al PrPSc original, pero al ser menor tamaño, ofrece más facilidades para el descifrado de su estructura.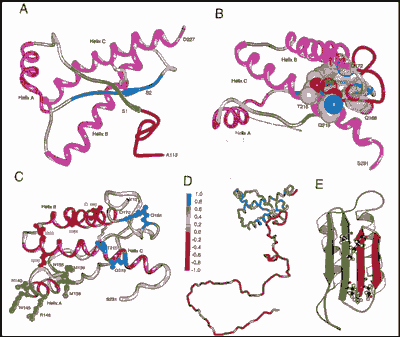
Miniprion creado borrando los residuos 23-89 y 141-176 de la PrP.
Arriba
La barrera de las especies.
La barrera fue descubierta por Pattison en los años 60. La causa de la barrera de las especies es la existencia de genes PRNP diferentes según las especies. Estos genes se traducirán en proteínas PrP con secuencias particulares, que constituirán una estructura terciaria de prion característica para cada especie.
La transmisión interespecífica de una enfermedad prion depende de que el prion patógeno del donante sea suficientemente similar a la proteína prion huésped como para poder unirse a ella e inducirle el plegamiento. El grado de compatibilidad de ambas proteínas determina el tiempo de incubación de la enfermedad (meses, años, décadas). Si las PrP son compatibles (como entre bovinos y humanos), no existe barrera interespecífica.
Como mejor se transmite la enfermedad (menor tiempo de incubación), es entre animales de la misma especie.
La estructura terciaria de las proteínas PrP es la que determina los linajes de los priones, (teoría apoyada inicialmente por la raza DY). Pueden distinguirse varios linajes (strains) de priones en una misma especie en función de tres estados de la proteína PrPSc:
- Sin glicosilar
- Monoglicosilada
- Biglicosilada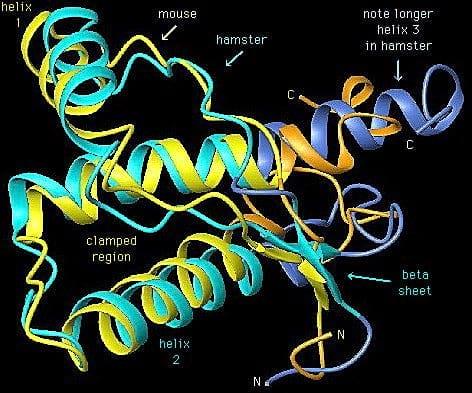
- Sin glicosilar
- Monoglicosilada
- Biglicosilada
El gen PrP del ratón difiere del de hámster en 16 codones de los 254 totales; y el de ratón del de humano en 28 codones, lo que da lugar a proteínas PrP con configuraciones distintas entre las especies.
Cuanto más se parezcan las secuencias PrPSc infecciosa y PrPc huésped, más probable será que el huésped adquiera la enfermedad prion. Se ha comprobado que el parecido en la región central de ambas moléculas, es más determinante que en otros segmentos.
Arriba
Arriba
La Proteína X
Es un pequeño ligando detectado mediante estudios genéticos y moleculares, que se une al PrPc y facilita su transformación a PrPSc.
Su descubrimiento se basó en la teoría según la cual, moléculas del huésped podían influir en el comportamiento del PrPSc. Para ratificar esta teoría se inocularon ratones transgénicos con genes PrP humanos (HuPrP) y con genes PrP híbridos entre ratón y humano (MHu2M). (6)
Los ratones con el gen híbrido infectados, desarrollaron la enfermedad más rápida y frecuentemente que los que poseían el gen totalmente humano. Este hecho llevó a afirmar la existencia de un factor procedente del ratón que reconocía las "regiones ratón" de la proteína PrP híbrida, facilitando el plegamiento de la PrPc híbrida; mientras que este factor no reconocía ninguna "región ratón" en la proteína PrPc totalmente humana (HuPrP) y, por tanto, no favorecía el proceso.
La proteína X está involucrada en el plegamiento de las proteínas a su forma PrPSc, y para ello debe reconocer ciertos fragmentos de la proteína PrP sobre la que actúa. De esta forma, priones PrPSc procedentes de especies muy alejadas evolutivamente de la del huésped al que infectan, no producirán la enfermedad fácilmente y necesitarán un larga periodo de incubación, ya que la proteína X no las reconocerá.
Por la función que ejercía esta proteína se pensó que podía tratarse de una chaperona, pero hasta el momento no se ha identificado ninguna chaperona molecular en mamíferos que intervenga en el proceso de plegamiento de priones.
Arriba
Diversidad de los priones.
Se observa una relación entre los patrones de deposición de PrPSc y los perfiles de vacuolación, y estos rasgos son usados para caracterizar las razas de priones.
Así, en la enfermedad prion FFI (fatal familiar insomnia) la deposición está confinada en gran parte al tálamo; en la enfermedad fCJD (familiar Creutzfeldt-Jakob Disease), los PrPSc se depositan en el manto cortical y en muchas de las estructuras profundas del SNC.
En la tabla siguiente se enumeran las enfermedades prion conocidas hasta ahora y alguna información nomenclatural referente a ellas:
Inicialmente, se pensaba que una mutación PrP específica estaba asociada con unos síntomas clínicos/neuropatológicos particulares. Pero ahora, con el reconocimiento de las enfermedades CJC, GSS y FFI familiares (es decir, heredadas) como autosómicas y dominantes, cada vez más, se han encontrado mutaciones PrP particulares en una familia que manifestaban síntomas clínicos diferentes a los acostumbrados.
Por ejemplo, muchos pacientes con la mutación PrP en el codón 102, presentaban ataxia y tenían placas amiloides PrP; estos pacientes eran diagnosticados como GSS, pero algunos de ellos desarrollaban la demencia propia del CJD.
Los priones de las vacas (Bos taurus) y ovejas (Ovis aries) son parecidos entre sí, ellos pertenecen a la familia de los Bovidae y comparten un antecesor común que vivió probablemente hace menos de 20 millones de años. Pero estos priones son muy distintos de los de humano (Homo), gorila (Gorilla), chimpancé (Pan) y el gran grupo de monos. 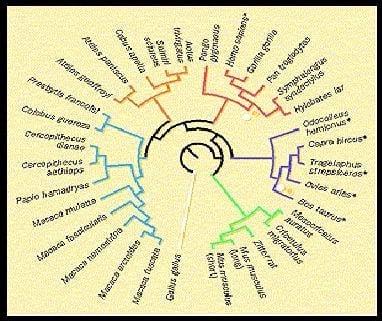
Árbol filogenético (Krakauer, 1996)
Las Vacas Locas o Síndrome de BSE
Las Vacas Locas o Síndrome de BSE
Según Según los vínculos observados, es lógico pensar que las vacas se puedan infectar de BSE, alimentándose de productos procedentes de ovejas enfermas con scrapies, pero no parece razonable que los humanos puedan enfermar de CJD por haberse alimentado de carne infectada con BSE.
El síndrome de BSE aparece en bovinos alimentados con productos derivados de ovejas, ganado, cerdos y pollos. Lo que permite que los priones de las ovejas enfermas de scrapie pasen a las vacas, produciendo la enfermedad BSE. Se ha observado que la secuencia PrP del bovino difiere en 7 u 8 posiciones de la de la oveja. Y, al contrario de los numerosos polimorfismos del PrP encontrados en la oveja, solo se ha hallado un polimorfismo PrP en los bovinos.
La probabilidad de que las especies Homo sapiens sapiens y Bos taurus, mantengan similaridades en sus moléculas PrP son muy remotas, pero estas, por improbables que sean, deben localizarse en las partes esenciales de transferencia de la enfermedad para que la enfermedad pueda saltar la barrera de las especies, tal y como sucede.
Se han descubierto dos pares de sustituciones que solo se encuentran en bovinos y hominoides, con excepción del orangután (la superfamilia Hominoidea incluye al hombre, chimpancé, gorila, gibón y orangután).
La sustituciones son la de Tyr que pasa a His en la posición 155 del gen PrP, y la de Asp a Ser en la 143. Quizá estas sustituciones tengan algún significado biológico, pero además predisponen a los humanos a estas enfermedades procedentes de los bovinos.
Como consecuencia de este síndrome de las vacas, en Gran Bretaña, apareció un caso particular de CJD que solo afectaba a adolescentes y adultos jóvenes, y que provocaba la aparición de numerosas placas PrP amiloides rodeadas por un halo de intensa degeneración espongiforme, se le llamó nvCJD (nueva variante). Los síntomas neuropatológicos observados en esta enfermedad, así como los patrones de glicoformas PrP observados en estos pacientes eran distintos de los hallados en las otras variantes de la enfermedad (sCJD, iCJD o fCJD). Tampoco se explica la predilección por individuos jóvenes (¿comer muchas hamburguesas?).
El BSE es identificado y se comienza a estudiar en los años 1985-86. La descripción del primer caso de nvCJD es 1996, y la confirmación de que la nvCJD es causada por el agente responsable del BSE es en 1997. Hasta ahora se han detectado 90 casos de nvCJD en toda la UE.
La sospecha inicial de que nvCJD procede del síndrome de las vacas locas viene de dos experimentos:
- La inoculación de tejido cerebral de BSE en monos produjo cambios clínicos y esponjiformes muy similares a los vistos en bovinos (Lasmezas 1996).
- El análisis de la electroforesis del amiloide extraido de las placas de nvECJ tiene un patrón más semblante al de BSE que al de ECJ esporádica.
Las sospechas se confirman en la publicación de Almond y Pattison en (Nature, 1997).
- La inoculación de tejido cerebral de BSE en monos produjo cambios clínicos y esponjiformes muy similares a los vistos en bovinos (Lasmezas 1996).
- El análisis de la electroforesis del amiloide extraido de las placas de nvECJ tiene un patrón más semblante al de BSE que al de ECJ esporádica.
Las sospechas se confirman en la publicación de Almond y Pattison en (Nature, 1997).
En Gran Bretaña se abolió en 1989 el uso de tejidos de alto riesgo como sesos y vísceras en la preparación de alimentos para el consumo humano. En España, las harinas animales están prohibidas para el vacuno desde 1994, pero han estado autorizadas para el porcino y el ovino hasta diciembre del 2000.
Los números del síndrome BSE hasta el año 2000:
- 177416 casos en Gran Bretaña, de los cuales 1801 en Irlanda del Norte.
- 538 casos en Irlanda.
- 458 casos en Portugal.
- 360 casos en Suiza.
- 176 casos en Francia.
- 24 casos de BSE en España hasta Febrero del 2001.
- 18 casos en Bélgica.
- 6 en Países Bajos.
- 6 en Alemania.
- 2 en Italia.
- 177416 casos en Gran Bretaña, de los cuales 1801 en Irlanda del Norte.
- 538 casos en Irlanda.
- 458 casos en Portugal.
- 360 casos en Suiza.
- 176 casos en Francia.
- 24 casos de BSE en España hasta Febrero del 2001.
- 18 casos en Bélgica.
- 6 en Países Bajos.
- 6 en Alemania.
- 2 en Italia.
Hasta Abril de 1999 se han detectado en la UE 179.087 casos de BSE, 99,5% de los cuales se han cuantificado en Gran Bretaña
Arriba
Nomenclatura.
Para simplificar la terminología se ha sugerido que la isoforma PrP que provoca la enfermedad, sea llamada PrPSc, sin mencionar el origen del prion. Para añadir especificidad un mutante o variante de PrP puede ser anotado entre paréntesis.
Por ejemplo: el prion encontrado en el ratón I/Ln, que posee una variante del gen PrP con F en el codon 108 y V en el 189, se identifica como MoPrP(108F, 189V)Sc; el prion encontrado en un judío Libio paciente de CJD, homozigótico para la mutación de K en el codón 200 se identificó como HuPrP(200K)Sc. En situaciones de heterozigosis y en las que el alelo que determina la forma PrP es desconocido, se usa HuPrPSc o HuPrPCJD o la enfermedad de la que se trate.
Arriba
Estructura y expresión del gen PrP.
El gen codificante del prion (llamado PRNP) se encuentra en el brazo corto del cromosoma 20. El PRNP normal contiene 5 octarepeticiones entre los codones 51 y 91; la ausencia de una octarepetición en el codon 81-82 no se asocia a ninguna enfermedad por priones. Inserciones adicionales de octarepeticiones se asocian con ECJ familiares, y determinadas mutaciones puntuales con otras enfermedades familiares por prion.
El gen PRNP es un único exon, no se han detectado intrones en ninguno de los genes PrP conocidos de mamíferos y aves, a excepción de los de ratones, ratas, ovejas y vacas, en los que se han encontrado tres intrones.
El mapeado comparado de los genes PrP del brazo corto del cromosoma 20 de los humanos y de la región homóloga del cromosoma 2 del ratón, aporta argumentos suficientes para pensar en la existencia de genes PrP anteriores a la especiación de mamíferos.
Aunque el ARNm del PrP se expresa únicamente en neuronas de animales adultos, está altamente regulado durante el desarrollo. Los niveles detectados más altos de ARNm PrP se encuentran en las neuronas.
La Proteína del anti-prion
La Proteína del anti-prion
El gen PRNP está muy conservado y tiene una sola pauta de lectura (ORF). Se ha encontrado otro ORF tan grande como el de PrPc en la hebra antisentido de las regiones codificantes del gen de mamíferos. También un ARNm de 4,5 kilobases (solo en tejido cerebral) del que se ha deducido una secuencia de aminoácidos reflejada a la de PrPc, lo que se ha llamado anti-PrP.
Se ha comprobado que este ARNm 4,5Kb procede de otro locus distinto del gen PRNP, pero a pesar de esto, codifica una proteína anti-prion que podría afectar la función normal de la proteína prion o también interactuar con ella en las células infectadas. Mutaciones encontradas en pacientes de GSS en los codones 102 y 117 del PRNP producían cambios en la secuencia primaria del anti-PrP, mientras que mutaciones encontradas en pacientes de CJD en los codones 178 y 200 no lo hicieron. (7 y 8)
Arriba
Prevención y tratamiento.
Actualmente es posible identificar las disfunciones neurológicas con mucha antelación, por lo que desarrollar un tratamiento para estas enfermedades es un imperativo, según Prusiner.
Para el diagnóstico de una demencia por priones debe observarse un cuadro clínico, neuropatológico y bioquímico compatible con una demencia rápidamente progresiva. No deben existir alteraciones metabólicas, lesiones estructurales cerebrales, infecciones ni tumores que puedan ser responsables de la encefalopatía y el deterioro mental.
El diagnóstico definitivo depende del examen directo del tejido cerebral por medio de una biopsia; puede ser confirmado mediante tinción histoquímica con anticuerpos contra la PrPSc en las placas de amiloide, también mediante el enzimoinmunoensayo, comprobando la presencia de anticuerpos contra PrP en Western Blot (Haywood 1997; Castellani 1996). Aunque los anticuerpos no son específicos contra PrPc o PrPSc, el tratamiento con proteinasa K elimina la PrPc.
La inmunohistoquímica puede detectar PrPc en secciones de tejido embebido en parafina, fijado con formalina y tratando con 90-100% de ácido fórmico de secciones desparafinadas.
Un estudio (Hisch 1996) demuestra especificidad y sensibilidad global de la proteína 14-3-3 en el diagnóstico de la ECJ esporádica en un 96%. Esta proteína es neuronal, está presente en diversas especies y podría jugar un papel importante en la estabilización de proteínas cerebrales en general. La sensibilidad de este examen se mantiene solo en ausencia de lesiones cerebrales agudas o subagudas ocurridas en el mes anterior al examen. Estudios posteriores parecen no coincidir con esta alta sensibilidad (Zeidler 1197a, Concha 1998).
Hay que decir que los casos estudiados que proporcionaron un 96% de sensibilidad son por lo general avanzados, en oposición a las etapas más tempranas de los pacientes vistos en la práctica clínica. Si la prueba es negativa inicialmente, se recomienda repetirla de 3 a 6 meses más tarde (CJ Gibbs).
El análisis de la concentración en suero de la proteína S100, específica de cerebro, puede ser una valiosa herramienta en el diagnóstico diferencial de CJD, más fácil de realizar que los test en fluido cerebroespinal. Este método de diagnóstico tiene una sensibilidad del 77.8% y una especifidad del 81,1% para confirmar el CJD. (13)
También es importante la exploración de mutaciones en el gen PRNP por su asociación con una transmisión autosómica dominante. Hasta hoy, las mutaciones conocidas tienen una penetrancia incompleta y, por tanto, la manifestación en portadores asintomáticos es incierta.
Igualmente, inmunoensayos para PrPSc en muestras cerebrales del ganado, pueden darnos una aproximación de cuanta incidencia de BSE de ganado está entrando en la cadena alimenticia humana.
Se precisan nuevos marcadores funcionales para diagnosticar de modo específico in vivo estas enfermedades. Herbert Budka del Instituto de Neurología de la Universidad de Viena, los busca en el SN periférico y afirma que el diagnóstico podría realizarse según "niveles determinados en la sangre". Él mismo coordina un estudio sobre las implicaciones epidemiológicas de la enfermedad por priones.
Pedro Piccardo, de la División de Neuropatología de la Universidad de Indiana, ha estudiado la enfermedad de Gerstmann-Straussler-Scheinker (GSS). Según él "hay una degradación distinta de la que clásicamente ha sido descrita en estas enfermedades", ello se debe a que algunos pacientes acumulan una isoforma de la proteína de bajo peso molecular. "Se considera como hipótesis que la proteína está implicada en el desarrollo de la enfermedad hereditaria", ha indicado. Piccardo ha diseñado "diferentes anticuerpos contra distintas regiones de la proteína, con la finalidad de describir su degradación".
La terapia de actuación más atractiva, es la que investiga como interferir la conversión de PrPc en PrPSc, estabilizando la estructura de la PrPc mediante la aplicación de un fármaco de unión. Falta por determinar si una droga capaz de ligarse a la PrPc en la zona de unión de la proteína X, puede ser más eficaz que otra que imite la estructura de la PrPc con residuos polimórficos básicos que parecen ser capaces de prevenir el scrapie y la enfermedad de Creutzfeldt-Jakob. Estos fármacos no precisan penetrar en el citosol de las células, aunque sí deben ser capaces de introducirse en el sistema nervioso central (SNC).
Otras líneas pueden ser:
- Búsqueda de drogas que desestabilicen la estructura de la PrPSc.
- Agentes que reduzcan la expresión del gen PrP y retrasen así la aparición de enfermedades de origen priónico.
- Productos químicos que interfieran la endocitosis, exocitosis, tráfico intracelular y degradación de proteínas y en particular, PrP.
- La anfotericina ha demostrado retrasar la enfermedad en hámsters (aunque aparentemente tiene poco efecto en humanos).
- La glicosilación puede modificar la conformación de la PrPc, afectando su afinidad por una forma particular de PrPSc, además, afecta a la estabilidad de la PrPSc.
- Dado que la muerte neuronal por la PrPsc se realiza mediante un proceso tóxico en el que se liberan moléculas oxidantes. Se ha experimentado satisfactoriamente con cultivos celulares en los que mediante la introducción de agentes antioxidantes como la Vit. E y la N-acetilcisteína se bloquea al efecto de la proteína infecciosa.
- Cría de animales que no sean capaces de replicar los priones. Ovejas que codifican el polimorfismo R/R en la posición 171 han demostrado ser resistentes al Scrapie. Una técnica más efectiva para producir animales de cría prion-resistentes, puede ser la expresión de transgenes PrP que codifiquen R117, así como residuos básicos adicionales para la zona de unión de la proteína X.
Igualmente, trata de determinarse si priones de otras proteínas intervienen en otros procesos neurodegenerativos como las enfermedades de Alzheimer, Parkinson, Huntington, Esclerosis lateral amiotrófica y otras.
- Búsqueda de drogas que desestabilicen la estructura de la PrPSc.
- Agentes que reduzcan la expresión del gen PrP y retrasen así la aparición de enfermedades de origen priónico.
- Productos químicos que interfieran la endocitosis, exocitosis, tráfico intracelular y degradación de proteínas y en particular, PrP.
- La anfotericina ha demostrado retrasar la enfermedad en hámsters (aunque aparentemente tiene poco efecto en humanos).
- La glicosilación puede modificar la conformación de la PrPc, afectando su afinidad por una forma particular de PrPSc, además, afecta a la estabilidad de la PrPSc.
- Dado que la muerte neuronal por la PrPsc se realiza mediante un proceso tóxico en el que se liberan moléculas oxidantes. Se ha experimentado satisfactoriamente con cultivos celulares en los que mediante la introducción de agentes antioxidantes como la Vit. E y la N-acetilcisteína se bloquea al efecto de la proteína infecciosa.
- Cría de animales que no sean capaces de replicar los priones. Ovejas que codifican el polimorfismo R/R en la posición 171 han demostrado ser resistentes al Scrapie. Una técnica más efectiva para producir animales de cría prion-resistentes, puede ser la expresión de transgenes PrP que codifiquen R117, así como residuos básicos adicionales para la zona de unión de la proteína X.
Igualmente, trata de determinarse si priones de otras proteínas intervienen en otros procesos neurodegenerativos como las enfermedades de Alzheimer, Parkinson, Huntington, Esclerosis lateral amiotrófica y otras.
Arriba
La herencia de la levadura.
En el caso de la levadura, el fenómeno de la herencia de los priones implica algo más importante que la simple transmisión de un agente infeccioso de las células madre a las hijas. Conlleva el traspaso de un rasgo genético particular.
Actualmente se sabe que ese rasgo genético se transmite por proteínas codificadas por el núcleo. Pueden heredarse mejor a través del citoplasma celular, que por medio de la información genética. Dichas proteínas pueden cambiar su conformación en el citoplasma, induciendo el mismo cambio a otras proteínas recién formadas.
Se han descrito en la levadura dos rasgos genéticos llamados [URE3] y [PSI]. Los responsables de los fenotipos [URE3] y [PSI] de la levadura, han sido identificados como los priones Ure2p y Sup35. Estos priones, en lugar de estar anclados en la membrana (mediante el ancla GPI), son citosólicos.
El rasgo [PSI], se sabe que controla la proporción de ribosomas que leen a través de los codones terminales. La proteína chaperona Hsp104 determina la herencia de este rasgo. En las células anormales [PSI+] se encontró el prion Sup35 con una conformación diferente a la que se encuentra en las células normales [psi-]. La forma alterada Sup35 induce el mismo cambio a las moléculas Sup35 recién sintetizadas, con la mediación de la HSP104. El rasgo [PSI+] proviene de la herencia de la proteína Sup35 en su forma alterada. (9 y 10)
Arriba
Arriba
El doble juego de una proteína.
Las HSP (heat shock protein) son chaperonas encargadas de proteger otras proteínas de síntesis reciente (no replegadas) o que se han desplegado después de un choque térmico, para evitar su interacción con otras moléculas. La producción de HSPs aumenta tras una elevación brusca de la temperatura, pero en concreto la HSP90, está constantemente presente en el citoplasma de las células eucariotas; y es que, en ausencia de estrés, esta proteína desarrolla otra función además de la de chaperona.
Las científicas Suzanne Rutherford y Susan Lindquist, de la Universidad de Chicago, estudiaron la descendencia de distintas poblaciones de Drosophila melanogaster a las que sometieron a mutaciones en el gen hsp90; alimentaron moscas genéticamente normales con una sustancia que inhibe la acción de la HSP90; y criaron moscas sanas en condiciones de choque térmico. El resultado en todos los casos fue el mismo: el rápido aumento de mutaciones en la descendencia, obteniendo entre el 80% y el 90% de las drosófilas anormales. La razón de esto es que las HSP90 habían mutado, estaban inhibidas o debían dedicarse a proteger a otras proteínas durante un estrés.
Rutherford y Lindquist propusieron que las mutaciones responsables de las anomalías morfológicas observadas estaban presentes en el patrimonio genético de las moscas antes de la manipulación que inactivaba a las HSP90. Según esta teoría, la HSP90 suprimiría los efectos que las mutaciones pudieran tener durante la embriogénesis. Así, permitiría la instalación de mutaciones genéticas silenciosas en una población sin que pudieran ser eliminadas por la selección natural. (9 y 10)
- Stanley B. Prusiner. Prions. Proceedings of the National Academy of Sciece of the USA. Vol. 95 - issue 23 - pp. 13363-13384
- (1) L. Manuelidis, T. Sklaviadis, A. Akowitz y W. Fritch. Viral particles are required for infection in neurodegenerative CJD disease. Proc. Natl. Acad. Sci. USA Vol. 92, pp. 5124-5128 (Mayo 1995)
- (2) Suehiro Sakaguchi y Cols. Loss of cerebellar Purkinje cells in aged mice homozigous for a disrupted PrP gene. Nature vol. 380: 528-531 (11 Abril/96)
- (3) S.B. Prusiner y Cols. Ablation of the prion protein (PrP) gene in mice prevents scrapie and facilitates production of anti-PrP antibodies. Proc. Natl. Acad. Sci. USA Vol.90, pp.10608-10612 (November 1993)
- (4) S. Mouillet-Richard, M. Ermonval, C. Chebassier, J.L. Laplanche, S. Lehman, J.M. Launay, O. Kellerman. Signal transduction trough prion protein. Science vol. 289: 1925-1927 (15 Septiembre/00)
- (5) Jeffrey Almond and John Pattison. Human BSE. Nature vol.389: 437-438 (2 Octubre/97)
- (6) G.C. Telling, M. Scott, K.K. Hsiao, etc.. Transmission of CJD disease from humans to transgenic mice expressing chimeric human-mouse prion protein. Proc. Natl. Acad. Sci. USA Vol. 91, pp. 9936-9940 (Octubre 1994)
- (7) Markus Moser, Bruno Oesch, Hansruedl Bueler Institut fur Hirnforschung Universitat Zurich. Nature Letter vol. 362: 213-214 (1993)
- (8) Goldgaber D. Nature vol. 351: 106 (1991)
- (9) Suzanne L. Rutherford and Susan Lindquist. Hsp 90 as a capacitor for morphological evolution. Nature vol. 396: 336-341 (26 Noviembre/98)
- (10) Mundo Científico vol. 204: 28-31 (Septiembre/99)
- (11) D.R. Brown, B.S. Wong, I.M. Jones. Recombinant prion protein exhibits superoxide dismutase. Journal of Neurochemistry 73, suppl. S19B
- (12) Lancet 2000; 356: 955-9
- (13) Markus Otto y cols. Diagnosis of CJD by measurement of S100 protein in serum: prospective case-control study. British Medical Journal 1998;316:577-582 ( 21 February )
- Conferencia de María Gasset. Centro Cultural Bancaja. Valencia, Enero del 2001
Arriba
Trabajo realizado por Carlos Sánchez Huertas, estudiante de Biología en la
Universidad de Valencia.
Universidad de Valencia.
Temas relacionados
Únete para poder comentar.
Una vez que te unas a Engormix, podrás participar en todos los contenidos y foros.
* Dato obligatorio
¿Quieres comentar sobre otro tema? Crea una nueva publicación para dialogar con expertos de la comunidad.
Crear una publicación
Instituto Nacional de Tecnología Agropecuaria - INTA
5 de septiembre de 2011
cynthia: dentro del Programa de Vigilancia de las Encefalopatías Espongiformes Transmisibles de los animales que se realiza en la Argentina, uno de sus pilares es la evaluación de las plantas que fabrican alimentos balanceados y determinar si existe contaminación cruzada de la producción de los mismo. Ver en página de SENASA la base del programa. Saludos
SENACSA Paraguay
28 de abril de 2011
Una de las principales hipotesis acerca de la transmición de esta enfermedad es por el consumo de balanceados infectado con proteina de carne y hueso elaborada de animales que murieron por esta enfermedad, me gustaria saber si existe algun tema sobre el control que realiza cada pais en sus balanceados y que tecnica laboratorial utilizan para el analisis de los ya mencionados balanceados


